





- Free Radical Polymerization Algorithm for a Thermoplastic Polymer Matrix : A Molecular Dynamics Study
Ji-Won Jung*, Chan-Wook Park*, Gun-Jin Yun*†
*Department of Mechanical & Aerospace Engineering, Seoul National University
*†Department of Mechanical & Aerospace Engineering, Seoul National University, Corresponding author (E-mail: gunjin.yun@snu.ac.kr)- 무정형 열가소성 고분자의 자유 라디칼 중합 분자동역학 시뮬레이션 알고리즘
정지원* · 박찬욱* · 윤군진*†
In this paper, we constructed a molecular dynamics (MD) polymer model of PMMA with 95% of conversion by using dynamic polymerization algorithm of a thermoplastic polymer based on free radical polymerization. In this algorithm, we introduced a united-atom level coarse-grained force field that combines the non-bonded terms from the TraPPE-UA force field and the bonded terms from the PCFF force field to alleviate the computation efforts. The molecular weight distribution and the average molecular weight of the polymer were calculated by investigating each chain generated from the free radical polymerization simulation. The molecular weight of the polymer was controlled by the number of initiator radicals presented in the initial state and molecular weight effect to the density, the glass transition temperature, and the mechanical properties were studied.
본 연구에서 우리는 자유 라디칼 중합에 기반한 열가소성 고분자의 동적 분자동역학 중합 알고리즘을 이용하여 95%의 변환률을 갖는 PMMA의 고분자 모델을 구성하였다. 본 알고리즘에서는 계산 수행에 필요한 시간을 줄이기 위해 PCFF 포텐셜 함수의 결합 항들 TraPPE-UA 포텐셜 함수의 비결합 항을 조합한 united-atom level의 coarse-grained 포텐셜 함수를 도입하였다. 자유 라디칼 중합 시뮬레이션을 통해 생성된 각 사슬을 분석하여 고분자의 분자량 분포와 평균 분자량을 계산하였고, 고분자의 분자량은 초기 상태에 존재하는 개시제 라디칼의 수를 이용하여 조절하였으며, 유리전이온도, 기계적 물성에 미치는 분자량의 효과에 대해 연구되었다.
Keywords: 자유 라디칼 중합(Free radical polymerization, 분자동역학(Molecular dynamics), 열가소성 고분자(Thermoplastic polymer), 열기계적 물성(Thermomechanical properties), PMMA(Poly methylmethacrylate)
최근 열가소성 고분자에 대한 연구를 통해 재료에 대한 이해가 높아짐에 따라 다양한 산업분야, 특히 자동차 및 항공우주 복합재료 분야에서 널리 이용되고 있다[1,2]. 열가소성 고분자는 재활용이 가능하고, 가벼우면서 제조 과정이 간단하여 비용이 절감될 뿐만 아니라 융합되고 높은 온도에서 용융된다는 특성 때문에 재성형이 가능하다는 측면에서 차세대 복합재료 기지로써 주목받고 있다. 최근 수십 년 사이에는 분자동역학 전산모사 기법(MD)이 발전하였으며, 실험 비용 절감과 분자단위에서의 고분자 현상의 이해를 위해 이용되어왔다.
MD 시뮬레이션은 고분자 연구 분야에서 유리전이온도(Tg), 영률, 밀도와 같은 열기계적 물성을 예측함으로써 그 신뢰성을 인정받아왔으며, 보다 정교한 분자 구조를 만들어 정확도를 향상시키고 시뮬레이션에 필요한 계산량을 줄이기 위한 연구가 지속되어왔다. 분자동역학에서고분자 모델을 구성하는 방식은 크게 두 가지: 동적 가교 방법[3-8]과 대표분자 방법[8-10]으로 분류할 수 있다. 동적 가교 방법은 임의로 에폭시 단량체와 가교제를 함께 채운 초기 상태에서 특정 거리 이내에 있는 활성 부위들을 결합시키는 방식을 통해 중합이 이루어지는 반면, 대표분자 방법은 초기에 단위 셀에 채워진 분자 상태에서 추가적인 결합이 이루어지지 않는다. 대신 특정 개수의 단량체들이 결합되어있는 대표분자 모델을 설정하여 단위 셀 내부에 복제되어 채우는 방식으로 이루어진다. 동적 가교 방법의 경우 주로 가교 네트워크 구조를 갖는 에폭시 수지의 모델링에 사용되었고, 가교가 존재하지 않고 사슬 구조를 갖는 열가소성 고분자에 대해서는 주로 대표분자 방법을 사용해왔다.
자유 라디칼 중합에 의해 중합되는 열가소성 고분자의 모델링에 동적 중합 방법을 이용한 기존 연구는 많이 이루어지지 않았다. Doherty et al.[11]는 치의학 분야에서 Bis-GMA/TEGMDA 구조의 사슬 성장에 대해 연구했다. Jang et al.[12]는 두 가지 종류의 vinyl ester(VE) 수지와 styrene의 공중합체에서의 사슬 성장에 대해 상대적 반응 부피의 개념을 적용한 연구를 하였지만, 사슬의 종결 반응에 대해서는 고려하지 않았다. 최근에 Torres et al.[13]는 hexanediol diacrylate(HDDA)의 가교 네트워크에 대하여 자유 라디칼 중합을 이용해 사슬의 종결반응을 포함한 연구를 진행하였으며, 고분자 구조의 토폴로지를 연구하여 고분자의 겔점을 구하고 80% 내외의 변환률에서 열기계적 물성을 계산하였다.
본 연구에서는 95%의 변환률을 갖는 poly(methyl methacrylate) (PMMA)의 구조를 자유 라디칼 중합 메커니즘에 기반한 동적 중합 방법을 이용해 구축하였다[14]. 시뮬레이션의 정확도를 잃지 않으면서 계산 시간을 줄이기 위해 united atom level의 coarse-grained 포텐셜을 이용하였으며, 단량체인 MMA 분자에 대한 개시제 라디칼의 비율을 1%, 3%, 5%로 변화시켜 평균 분자량을 조절시키면서 PMMA 구조의 열기계적 물성에 평균 분자량이 미치는 영향을 조사하였다.
자유 라디칼 중합은 총 3개의 단계: 개시, 성장, 정지로 이루어져있다. 개시 단계는 개시제 라디칼의(I·) 생성과 한 개의 단량체와의 결합을 포함한다. 개시제 라디칼이 생성된 이후에 주변의 π결합을 공격하여 라디칼과 단량체 사이에 공유결합을 형성하고, 라디칼은 단량체의 끝으로 이동하면서 이중결합은 단일결합으로 변환된다. (I· + M → IM·) 성장 반응은 개시 단계에서 설명된 것과 같은 방식으로 새로운 단량체가 사슬의 말단에 첨가되는 방식으로 진행된다(IMn-1· + M → IMn·). 종결 반응은 두 사슬의 말단 부위가 서로 만날 때 발생하며, 라디칼들의 비가역적인 소멸이 일어나 사슬의 성장이 멈추게 된다. 종결 반응에는 조합 반응과 불균등화 반응의 두 가지가 존재하며, 조화 반응이 일어날 경우 두 사슬 말단이 결합하여 하나의 사슬이 되고,(Mn· + Mm· →Mm+n) 불균등화 반응이 일어날 경우 두 개의 죽은 사슬이 생성된다(Mn·+ Mm· → Mn+ Mm).
본 연구에서의 PMMA 분자동역학 모델의 구성은 Materials Studio 2017 R2 package를 사용하였다[15]. 개시제로는 phenyl radical을 사용하였으며, 중합의 개시 단계는 개시제 라디칼과 함께 단량체를 단위 셀에 채우는 방식으로 이루어졌다. 사슬의 성장은 MMA단량체에 포함된 C=C 결합이 단일결합으로 변환되면서 진행된다. 사슬의 성장은 머리에서 꼬리 방향(이중결합을 이루는 두 개의 탄소 원자 중 기능기가 결합된 원자를 머리, 나머지를 꼬리라 칭한다.)으로 이루어진다고 가정하였으며[16], 공유결합은 각 성장 단계에서 cutoff radius 이내의 라디칼과 꼬리 원자가 결합되도록 하였다. 그 후 이중결합은 단일결합으로 변환되고 꼬리 원자는 새로운 라디칼 원자가 된다. 결합 이후에는 기존의 라디칼 원자는 비활성화되고, 각 성장 단계마다 2000step의 포텐셜 에너지 최소화 과정을 통해 인위적인 결합과정을 보상하였다. 각 과정은 cutoff radius 이내의 활성 라디칼과 머리 원자 쌍을 찾을 수 없을 때까지 반복되며, 더 이상 원자 쌍이 존재하지 않을 경우 360 K과 1 atm의 조건에서 NPT (일정한 개수의 입자, 압력, 온도)앙상블을 1 femtosecond(fs)의 time step으로 2 picoseconds(ps)동안 수행한다. 그 이후 cutoff radius를 7 Å에서 11 Å까지 0.5 Å씩 증가시키며 위의 과정을 반복 수행한다.
초기상태의 중합이 진행되지 않은 고분자의 단위 셀에는 6000개의 MMA단량체가 포함되었으며, 개시제 분자의 개수를 조절하여 평균 분자량이 서로 다른 3개의 모델을 구성하였다. 각 모델에는 단량체 분자에 대한 개시제 분자의 비율을 1%, 3%, 5%로 조절하여 각각 60, 180, 300개의 개시제 분자가 포함되었다.
본 연구에서는 각 원자간의 포텐셜을 계산하기 위해 조합된 PCFF-TraPPE(UA) 포텐셜 함수를 도입하였다[17]. 계산에 소모되는 시간을 절감하기 위해 탄소 원자와 그 주변에 결합된 수소 원자를 한 개의 입자로 계산하는 united atom level의 coarse-grained 모델이 적용되었다. PCFF-TraPPE(UA) 포텐셜은 PCFF 포텐셜의 결합 항과 TraPPE(UA) 포텐셜의 비결합 항을 조합함으로써 구성되었다. 연구에 사용한 MMA단량체와 개시제의 모델과 포텐셜은 Fig. 1에 표시되어 있다.
본 연구에서 PCFF의 결합항을 사용한 이유는 TraPPE(UA)에 묘사된 결합 포텐셜 함수의 형태는 2차항의 형태를 갖고 있기 때문으로[17], 이 경우 고분자 모델을 크게 변형시켜 입자 간 결합 길이가 크게 변형될 경우 인력이 과도하게 크게 계산된다[18]. 반면에, PCFF 포텐셜의 결합 항의 경우에는 4차 함수의 형태를 갖고 있기 때문에, 보다 정교하게 결합 길이에 따른 입자 간의 힘을 계산할 수 있다[19].
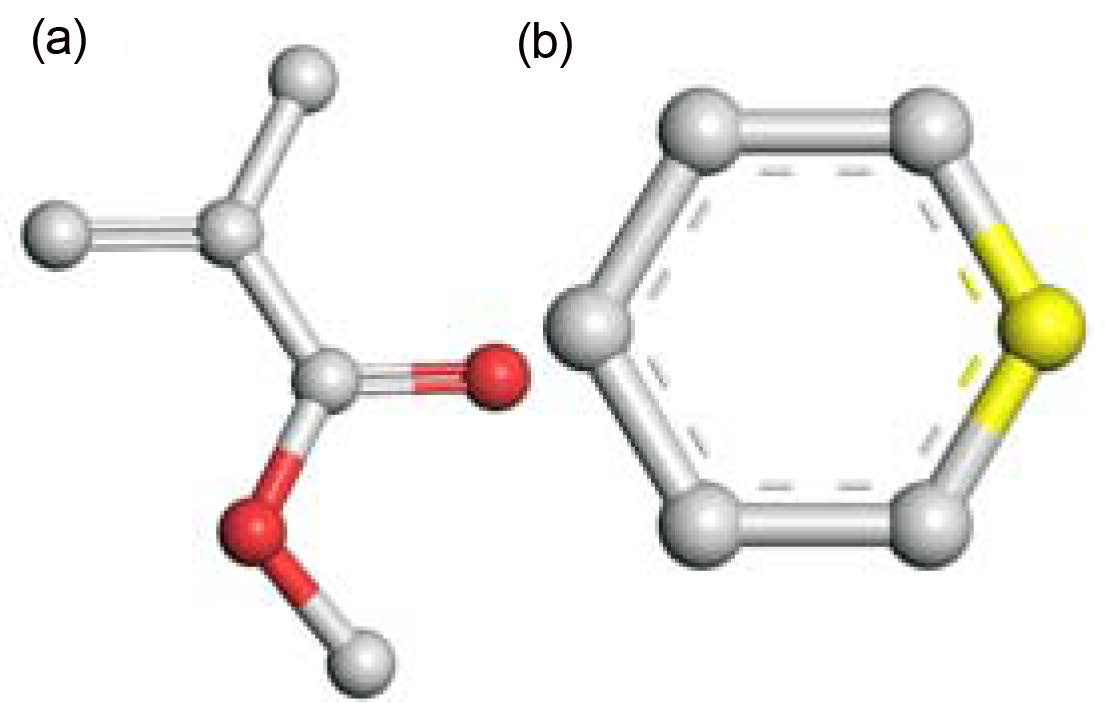
|
Fig. 1 The united atom level coarse-grained representation of a MMA monomer (a) and an initiator (b) where the white atoms are united carbon, the red atoms are oxygen and the yellow atom is the radical site in the initiator |
자유 라디칼 중합 알고리즘을 통해 생성된 고분자 구조에 대해 large-scale atomic/molecular massively parallel simulator(LAMMPS)[20] 시뮬레이터를 이용하여 유리전이온도와 기계적 물성을 계산하였다. 유리전이온도는 실험과 동등한 방법인 cooling down 시뮬레이션을 통해 구하였다. 고분자 구조를 cooling down하는 과정에서 온도에 따른 부피 변화의 기울기가 변하는 지점을 고분자의 상변화가 일어나는 온도로 가정하였다. Cooling down 시뮬레이션은 고온의 600 K에서 저온의 200 K까지 1 × 1011 K/s의 cooling rate로 수행되었다.
기계적 물성은 고분자 구조에 대한 단축 인장 시뮬레이션과 전단 시뮬레이션을 수행함으로써 측정되었다. 고분자 모델은 등방성의 재료 특성을 갖는다고 가정하여 x방향으로의 단축 인장 시뮬레이션과 xy방향의 전단 시뮬레이션을 수행하였으며, x방향으로의 strain rate와 xy방향으로의 shear strain rate는 2 × 109 /s로 설정하였다. x방향의 인장 시뮬레이션에서 변형이 일어나지 않는 다른 두 방향에 대해서는 푸아송 효과를 고려하기 위해 1기압과 300 K 조건에서의 NPT 앙상블이 적용되었으며, 전단 시뮬레이션에서는 300 K에서의 NVT 앙상블이 적용되었다. 인장 시뮬레이션과 전단 시뮬레이션은 모두 14%의 변형률까지 수행되었으며, 선형구간에서의 기울기를 통해 재료의 기계적 물성을 계산하였다.
고분자는 각각 사슬의 중합도에 차이를 갖고 있기 때문에 서로 다른 분자량을 갖고 있다. 이러한 고분자의 특성 때문에, 고분자의 분자량은 분자량 분포로써 묘사되며, 이는 평균분자량과 분산도로 표현된다. 고분자의 분자량은 고분자의 특성에 직접적인 영향을 미치기 때문에 고분자의 분자량을 연구하는 것이 중요하다.
그러나, 대부분의 분자동역학을 이용한 고분자의 연구에서 분자량에 대한 효과는 고려되지 않았다. 가교 결합이 존재하는 에폭시 고분자의 경우 거의 대부분의 고분자들이 가교결합을 통해 서로 연결되어있다. 그에 따라 분자량이 거의 무한에 가깝기 때문에 분자량에 의한 효과를 크게 고려하지 않았다. 반면에, PMMA 또는 polystyrene과 같은 열가소성 고분자의 경우에는 유한한 크기의 분자량을 갖기 때문에 분자량에 의한 효과를 고려해야 한다. 분자량을 대표하기 위한 평균분자량으로는 수 평균 분자량(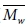 )과 무게 평균 분자량(
)과 무게 평균 분자량(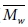 )을 흔히 이용하며 그에 대한 정의는 다음과 같다.
)을 흔히 이용하며 그에 대한 정의는 다음과 같다. 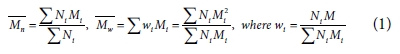
수식에서 표시된 Ni는 각 분자량에 해당하는 고분자 사슬의 개수, Mi는 사슬의 분자량을 뜻한다. 분자량 분포의 산포도는 다분산지수(poly dispersity index, PDI)를 통해 나타나며, 이는 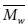 에 대한
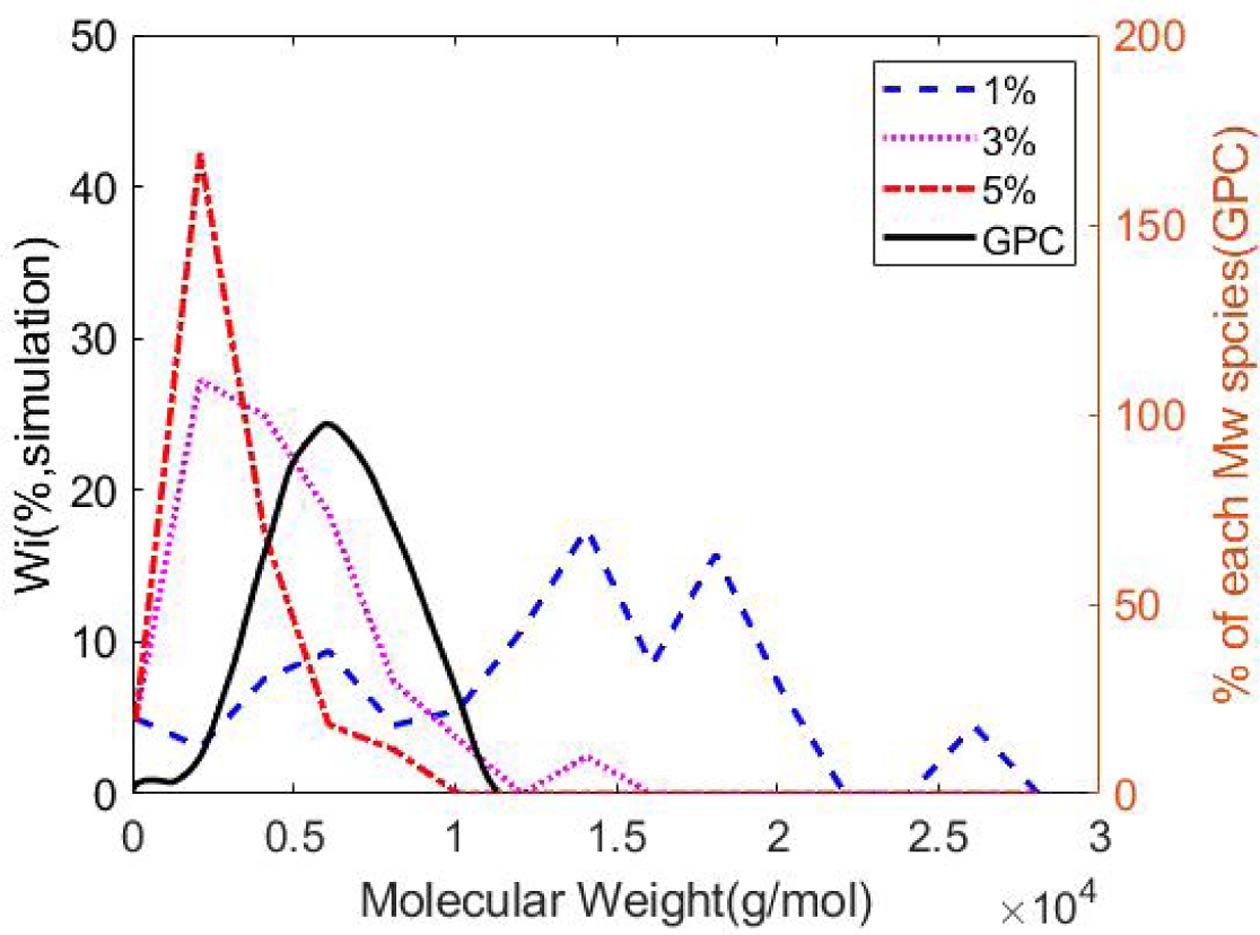
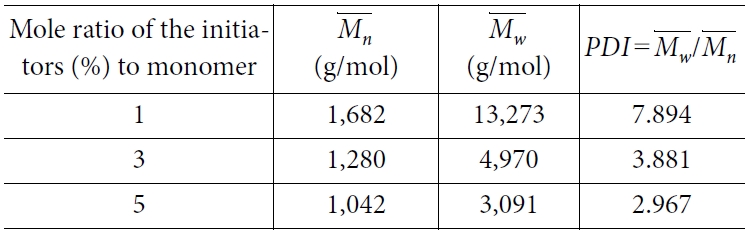
에 대한 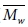 의 비율로써 정의된다. 분자량 분포가 균일한 분포를 가질수록 PDI 값은 1에 수렴하게 되며, 더욱 분산된 분포를 가질수록 PDI 값은 커진다. 자유 라디칼 중합을 통해 생성된 PMMA구조의 분자량 분포는 각 사슬의 길이를 분석함으로써 구했고, 그 분포도는 각 사슬의 분자량에 따른 무게 분율(wi)의 그래프로 그렸다. 평균 분자량과 분자량 분포도는 각각 Table 1과 Fig. 2에 표시되어있다. 그 결과 자유 라디칼 중합 알고리즘을 통해 생성된 분자동역학 모델의 분자량 분포가 실험에서 얻어지는 분포와 유사한 형태를 나타내는 것으로 보여졌다.
의 비율로써 정의된다. 분자량 분포가 균일한 분포를 가질수록 PDI 값은 1에 수렴하게 되며, 더욱 분산된 분포를 가질수록 PDI 값은 커진다. 자유 라디칼 중합을 통해 생성된 PMMA구조의 분자량 분포는 각 사슬의 길이를 분석함으로써 구했고, 그 분포도는 각 사슬의 분자량에 따른 무게 분율(wi)의 그래프로 그렸다. 평균 분자량과 분자량 분포도는 각각 Table 1과 Fig. 2에 표시되어있다. 그 결과 자유 라디칼 중합 알고리즘을 통해 생성된 분자동역학 모델의 분자량 분포가 실험에서 얻어지는 분포와 유사한 형태를 나타내는 것으로 보여졌다.
각 개시제의 비율에 따라 분자량 분포와 평균 분자량을 조사한 결과 낮은 개시제의 비율이 포함되었을 때 평균 분자량이 높은 것으로 나타났다. 이는 초기 모델에 포함된 단량체 분자의 개수와 변환률이 같아 중합 반응에 참여하는 단량체 분자의 개수가 같기 때문이다. 각 사슬은 개시제로부터 성장을 시작하기 때문에 사슬의 개수는 개시제의 수와 같고, 따라서 개시제가 적을수록 각 사슬에 결합된 단량체의 개수가 평균적으로 많아지는 것으로 해석된다. 또한 낮은 개시제 비율의 모델에서 다분산지수가 높아지는 현상을 확인했는데, 이러한 현상은 분자량이 작은 단량체들이 분자량 계산에 포함되어있기 때문으로, 평균 분자량과 단량체의 분자량 차이가 더 크기 때문에 분산이 높게 나타난 것으로 해석된다.
|
Fig. 2 Molecular weight distribution of the PMMA model of 1%, 3%, 5% initiator ratio plotted with typical experimental distribution of PMMA [21]. The molecular weight distribution diagram from the experimental result was rescaled to visually compare to simulation results |
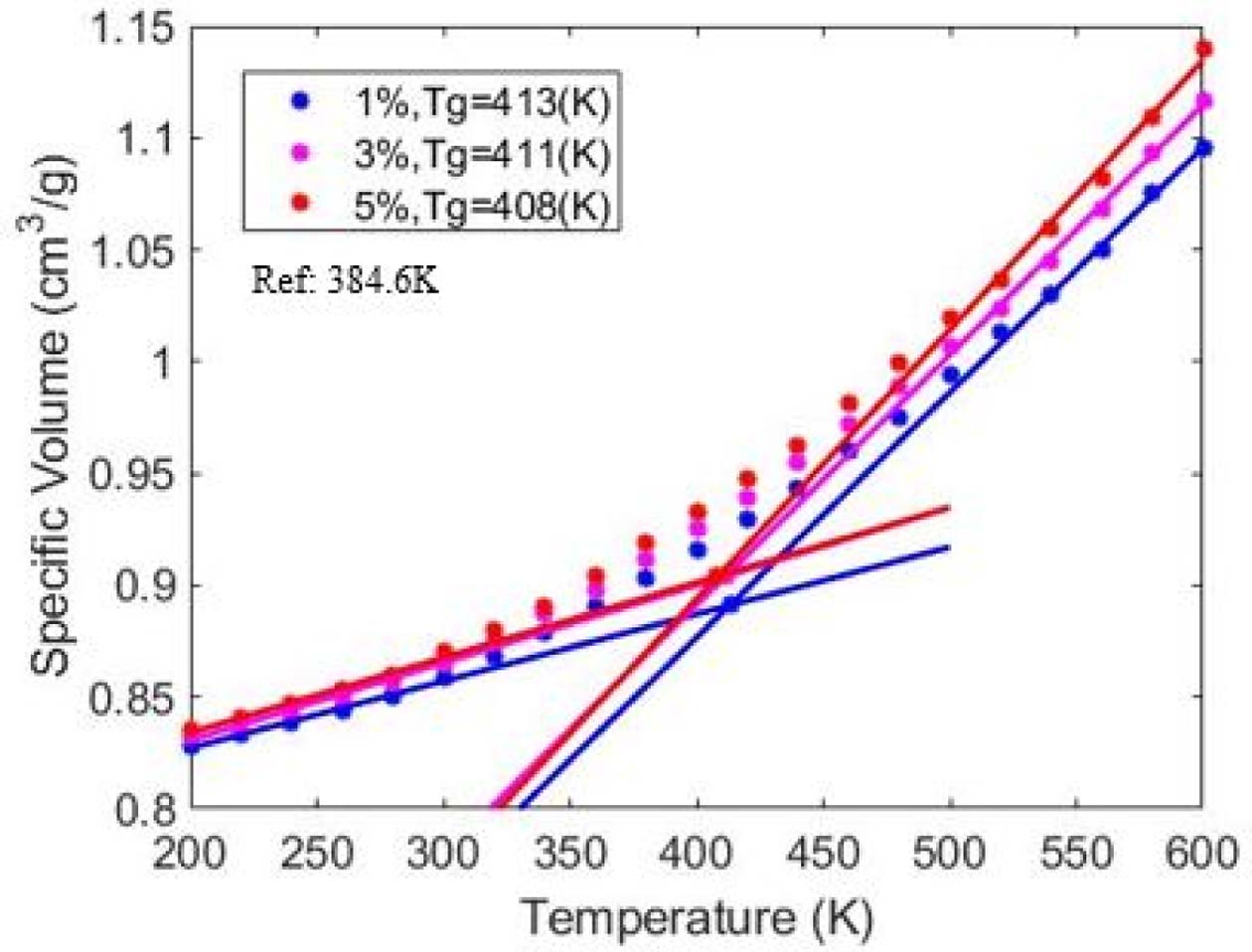
PMMA의 Tg는 413 K~408 K의 값을 갖는 것으로 계산되었다. 이러한 값의 범위는 실험을 통해 알려진 값인 384.6 K보다 높은 값이며, 이러한 차이는 MD 시뮬레이션이 갖고 있는 시간 단위에 따른 실험과의 cooling rate의 차이 때문이다[22]. 각 고분자 모델의 Tg는 분자량이 감소함에 따라 낮아지는 것을 Fig. 3을 통해 확인할 수 있었으며, 이러한 관계는 실험에 의해 다음과 같은 식 (2)로 표현된다[23,24].

식에서 나타난 는 분자량이 무한이라고 가정했을 때의 T∞g 값이며, K는 고분자의 종류에 따른 상수 값이다. 고분자의 Tg 값의 변화는 고분자의 자유부피의 변화에 따른 결과로 이해할 수 있다[16,24]. 자유 부피는 분자 사이의 비어있는 공간으로 정의되며, Tg 아래의 온도에서 유리상태에서는 일정한 값을 갖고, Tg 이상의 고무상에서는 온도가 감소함에 따라 자유부피가 감소한다. 낮은 분자량의 고분자 구조에서는 상대적으로 큰 자유부피를 차지하는 사슬 말단이 더욱 많이 존재하고, 그에 따라 유리상태에 도달하기 위해서는 더욱 큰 자유부피의 감소가 필요하기 때문에 낮은 온도에서 상변화가 일어나게 된다.
|
Fig. 3 Glass transition temperature of PMMA for different initiator to monomer ratio |
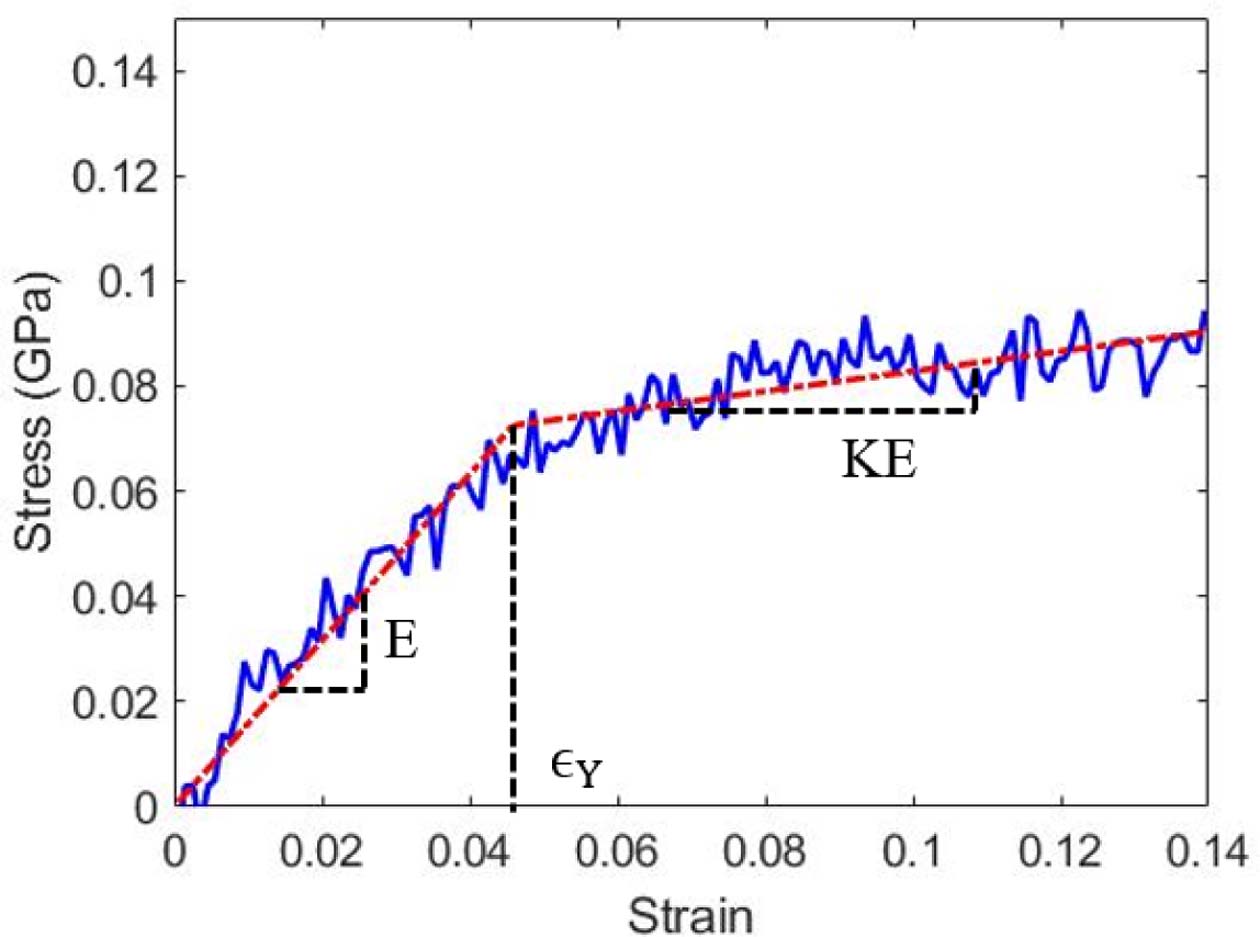
고분자의 단축 인장 시뮬레이션과 전단 시뮬레이션을 통해 얻어진 응력-변형률 곡선의 선형구간에서의 기계적 강성과 재료의 거동이 소성 영역에 도달하는 항복 응력을 도출하였다. 각 모델에 대해 인장 및 전단 시뮬레이션은 3회씩 수행하였으며, 응력-변형률 곡선은 bilinear elasto-plastic model을 이용하여 피팅하였고[25], 그 수식은 다음 식 (3)과 Fig. 4와 같이 표현된다.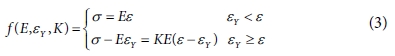

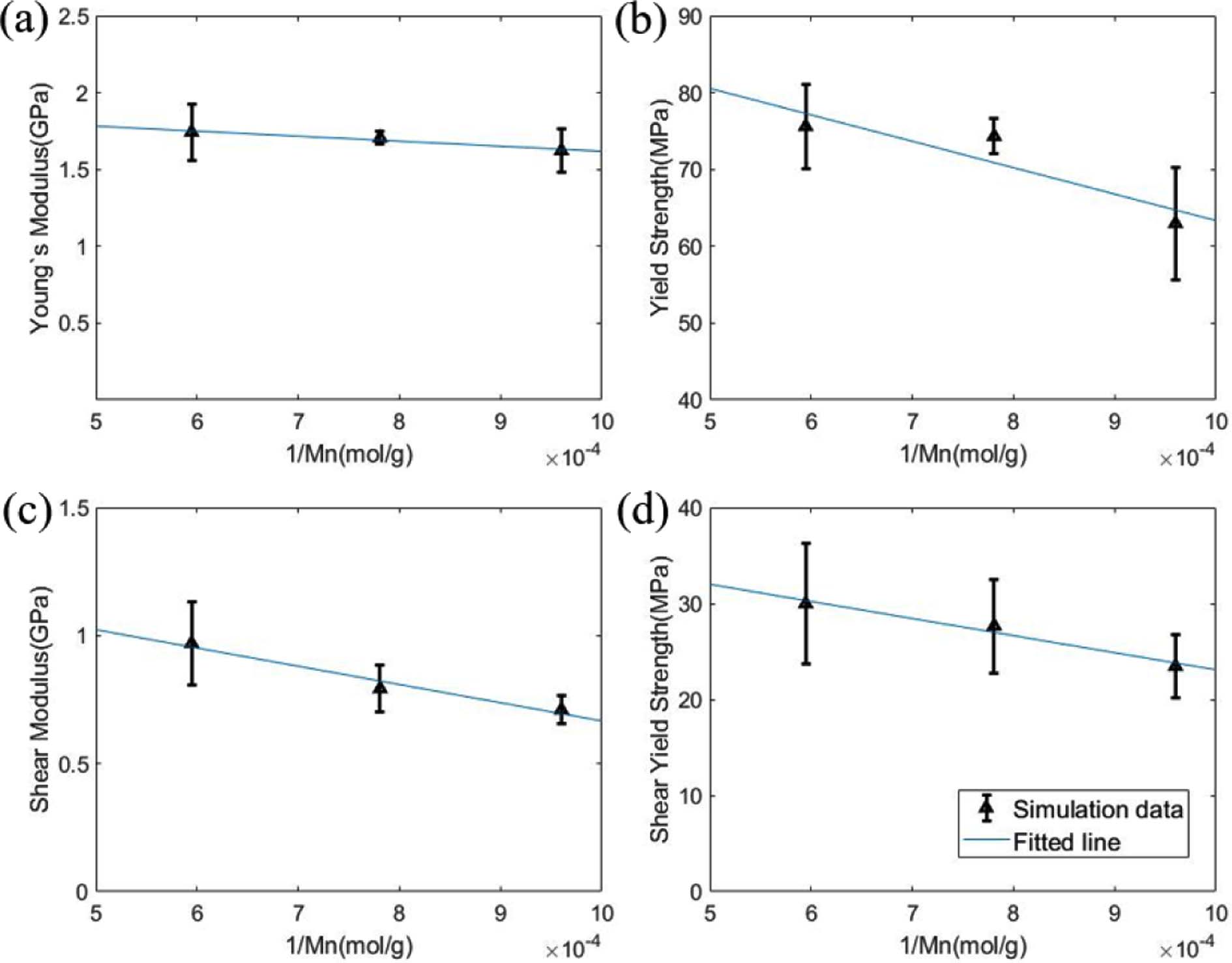
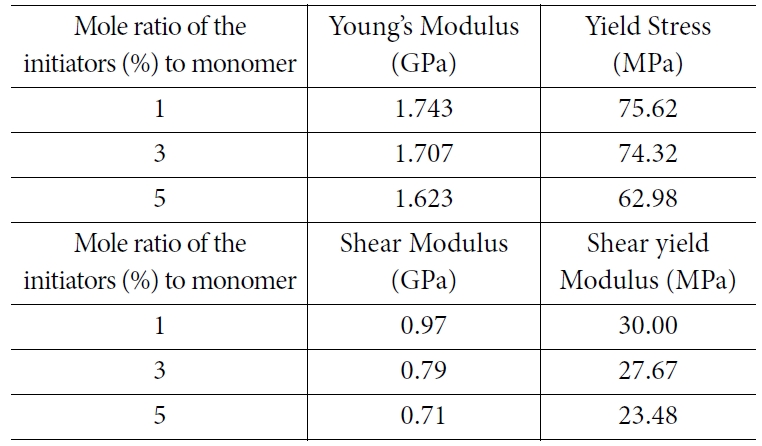
식 (3)에 표현된 bilinear model은 항복 스트레인 이전의 탄성구간과 항복 스트레인 이후의 소성구간을 나누어 2개의 선형 구간으로 나타난다. Bilinear 모델에서 피팅하여 결정하는 파라미터는 3개로 탄성구간에서의 기울기인 영률(E), 기울기가 바뀌는 지점인 항복 스트레인(εY)과 소성구간과 탄성구간의 기울기 비율인 hardening parameter(K)로, 시뮬레이션을 통해 얻은 응력-변형률 선도와 bilinear model간의 오차가 최소가 되도록 하는 방법을 통해 결정하였다. 위와 같은 피팅 방법을 통해 각 분자량에 따라 인장 및 전단 강성과 항복 응력을 비교하여 Fig. 5와 Table 2에 표시하였다.
시뮬레이션 결과 분자량이 증가함에 따라 기계적 물성이 증가하는 경향이 있음을 확인할 수 있었다. 유리전이 온도 아래의 무정형 고분자에서 발생하는 이러한 현상은 기존의 실험에 의해 보고된 바 있으며, 그 원인은 고분자 사슬간의 얽힘의 증가로 해석된다[26]. 이러한 물성의 증가효과는 특정 분자량 까지만 나타나며, (PMMA의 경우에는  <1.5x105) 그 이후에는 분자량이 증가해도 얽힘이 증가하지 않기 때문에 물성의 증가가 나타나지 않는다.
<1.5x105) 그 이후에는 분자량이 증가해도 얽힘이 증가하지 않기 때문에 물성의 증가가 나타나지 않는다.
|
Fig. 4 Bilinear elasto-plastic model for fitting SS-curve |
|
Fig. 5 (a) Young’s modulus, (b) tensile yield strength, (c) shear modulus, (d) shear yield strength plotted by the reciprocal number of molecular weights |
본 연구에서는 자유 라디칼 중합 분자동역학 알고리즘을 통한 PMMA구조를 구축하였으며, 알고리즘 상에서의 초기 모델의 개시제 농도 조절을 통해 서로 다른 분자량의 PMMA 모델을 얻어냈다. 자유 라디칼 중합 알고리즘을 통해 얻은 고분자 모델은 기존의 일정한 길이의 사슬을 복제하여 모델이 구성되는 대표분자 방법과 비교하여 실제 구조와 유사한 형태의 분자량 분포를 갖는 현상을 확인할 수 있었다. 또한 각기 다른 분자량을 갖는 3개의 모델에 대하여 유리전이온도와 기계적 물성을 계산하였으며, 그 결과 고분자의 열기계적 물성이 실험과 일치하는 경향성을 보임을 확인하였다. 이를 통해 자유 라디칼 중합 알고리즘을 통해 생성된 분자동역학 모델이 고분자 사이의 자유부피 및 사슬 간 얽힘과 같은 고분자의 특성들을 잘 반영하고 있음을 알 수 있다.
이러한 실제적 특성을 반영한 분자동역학 고분자 모델의 구성 연구는 열가소성 고분자 기지를 포함하는 복합재료 연구에 있어 분자간의 얽힘과 분자량 분포의 효과를 고려할 수 있기 때문에, 다른 filler가 함께 포함되어 적용된다면 열가소성 고분자가 포함된 복합재료 연구에 있어 기초적인 연구로써 활용될 수 있을 것으로 예상된다.
본 연구는 한국연구재단 주관 중견 연구자 지원 사업(2017R1A2B40004996) 연구과제로 수행된 것이며, 지원에 대해 진심으로 감사드립니다.
- 1. Takeuchi, K., Matsuzaki, R., Okabe, T., and Oya, Y., “Process Design for Heat Fusion of Thermoplastic Composites Using Molecular Dynamics and a Response Surface Method,” Advanced Composite Materials, Vol. 25, No. sup1, 2016, pp. 33-49.
-
- 2. Matsuo, T., Goto, T., and Takahashi, J., “Investigation About the Fracture Behavior and Strength in a Curved Section of CF/PP Composite by a Thin-curved Beam Specimen,” Advanced Composite Materials, Vol. 24, No. 3, 2015, pp. 249-268.
-
- 3. Wu, C., and Xu, W., “Atomistic Molecular Modelling of Crosslinked Epoxy Resin,” Polymer, Vol. 47, No. 16, 2006, pp. 6004-6009.
-
- 4. Varshney, V., Patnaik, S.S., Roy, A.K., and Farmer, B.L., “A Molecular Dynamics Study of Epoxy-Based Networks: Cross-Linking Procedure and Prediction of Molecular and Material Properties,” Macromolecules, Vol. 41, No. 18, 2008, pp. 6837-6842.
-
- 5. Bandyopadhyay, A., Valavala, P.K., Clancy, T.C., Wise, K.E., and Odegard, G.M., “Molecular Modeling of Crosslinked Epoxy Polymers: The Effect of Crosslink Density on Thermomechanical Properties,” Polymer, Vol. 52, No. 11, 2011, pp. 2445-2452.
-
- 6. Khare, K.S., and Khare, R., “Directed Diffusion Approach for Preparing Atomistic Models of Crosslinked Epoxy for Use in Molecular Simulations,” Macromolecular Theory and Simulations, Vol. 21, No. 5, 2012, pp. 322-327.
-
- 7. Hadden, C.M., Jensen, B.D., Bandyopadhyay, A., Odegard, G.M., Koo, A., and Liang, R., “Molecular Modeling of EPON-862/graphite Composites: Interfacial Characteristics for Multiple Crosslink Densities,” Composites Science and Technology, Vol. 76, No. 2013, pp. 92-99.
-
- 8. Park, C., and Yun, G.J., “Characterization of Interfacial Properties of Graphene-Reinforced Polymer Nanocomposites by Molecular Dynamics-Shear Deformation Model,” Journal of Applied Mechanics, Vol. 85, No. 9, 2018, pp. 091007.
-
- 9. Yu, S., Yang, S., and Cho, M., “Multi-scale Modeling of Cross-linked Epoxy Nanocomposites,” Polymer, Vol. 50, No. 3, 2009, pp. 945-952.
-
- 10. Park, C., Jung, J., and Yun, G., “Interfacial Characterization of Mineralized Carbon Nanotubes,” Composites Research, Vol. 31, No. 5, 2018, pp. 282-287.
-
- 11. Doherty, D., Holmes, B., Leung, P., and Ross, R., “Polymerization Molecular Dynamics Simulations. I. Cross-linked Atomistic Models for Poly(methacrylate) Networks,” Computational and Theoretical Polymer Science, Vol. 8, No. 1-2, 1998, pp. 169-178.
-
- 12. Jang, C., Lacy, T.E., Gwaltney, S.R., Toghiani, H., and Pittman, C.U., “Relative Reactivity Volume Criterion for Cross-Linking: Application to Vinyl Ester Resin Molecular Dynamics Simulations,” Macromolecules, Vol. 45, No. 11, 2012, pp. 4876-4885.
-
- 13. Torres-Knoop, A., Kryven, I., Schamboeck, V., and Iedema, P.D., “Modeling the Free-radical Polymerization of Hexanediol Diacrylate (HDDA): A Molecular Dynamics and Graph Theory Approach,” Soft Matter, Vol. 14, No. 17, 2018, pp. 3404-3414.
-
- 14. Jung, J., Park, C., and Yun, G.J., “Free Radical Polymerization Simulation and Molecular Entanglement Effect on Large Deformation Behavior,” European Polymer Journal, Vol. 114, No. 2019, pp. 223-233.
-
- 15. Dassault Systèmes BIOVIA, Materials Studio, 2017 R2, San Diego: Dassault Systèmes, 2017.
- 16. Young, R.J., and Lovell, P.A., Introduction to Polymers. CRC Press, 2011.
- 17. Maerzke, K.A., Schultz, N.E., Ross, R.B., and Siepmann, J.I., “TraPPE-UA Force Field for Acrylates and Monte Carlo Simulations for Their Mixtures with Alkanes and Alcohols,” The Journal of Physical Chemistry B, Vol. 113, No. 18, 2009, pp. 6415-6425.
-
- 18. Nouri, N., and Ziaei-Rad, S., “A Molecular Dynamics Investigation on Mechanical Properties of Cross-Linked Polymer Networks,” Macromolecules, Vol. 44, No. 13, 2011, pp. 5481-5489.
-
- 19. Sun, H., Mumby, S.J., Maple, J.R., and Hagler, A.T., “An ab initio CFF93 All-atom Force Field for Polycarbonates,” Journal of the American Chemical Society, Vol. 116, No. 7, 1994, pp. 2978-2987.
-
- 20. Plimpton, S., “Fast Parallel Algorithms for Short-range Molecular Dynamics,” Journal of Computational Physics, Vol. 117, No. 1, 1995, pp. 1-19.
-
- 21. Lehrle, R.S. and Sarson, D.S., “Polymer Molecular Weight Distribution: Results from Matrix‐assisted Laser Desorption Ionization Compared with Those from Gel‐permeation Chromato- graphy,” Rapid Communications in Mass Spectrometry, Vol. 9, No. 1, 1995, pp. 91-92.
-
- 22. Mohammadi, M., fazli, H., Karevan, M., and Davoodi, J., “The Glass Transition Temperature of PMMA: A Molecular Dynamics Study and Comparison of Various Determination Methods,” European Polymer Journal, Vol. 91, No. 2017, pp. 121-133.
-
- 23. Beevers, R., and White, E., “Physical properties of vinyl polymers. Part 1.—Dependence of the Glass-transition Temperature of Polymethylmethacrylate on Molecular Weight,” Transactions of the Faraday Society, Vol. 56, No. 1960, pp. 744-752.
-
- 24. Fox Jr, T.G., and Flory, P.J., “Second‐order Transition Temperatures and Related Properties of Polystyrene. I. Influence of Molecular Weight,” Journal of Applied Physics, Vol. 21, No. 6, 1950, pp. 581-591.
-
- 25. Park, H., Choi, J., Kim, B., Yang, S., Shin, H., and Cho, M., “Toward the Constitutive Modeling of Epoxy Matrix: Temperature-accelerated Quasi-static Molecular Simulations Consistent with the Experimental Test,” Composites Part B, Vol. 142, No. 1, 2018, pp. 131-141.
-
- 26. Martin, J.R., Johnson, J.F., and Cooper, A.R., “Mechanical Properties of Polymers: The Influence of Molecular Weight and Molecular Weight Distribution,” Journal of Macromolecular Science, Part C, Vol. 8, No. 1, 1972, pp. 57-199.
-
 This Article
This Article
-
2019; 32(3): 163-169
Published on Jun 30, 2019
- 10.7234/composres.2019.32.3.163
- Received on Mar 25, 2019
- Revised on Jun 27, 2019
- Accepted on Jun 28, 2019
Services
Shared
Correspondence to
- Gun-Jin Yun
-
Department of Mechanical & Aerospace Engineering, Seoul National University
- E-mail: gunjin.yun@snu.ac.kr
Gangnam Mirae Tower, Suite 601, 174 Saimdang-ro, Seocho-gu, Seoul 06627, South Korea
Tel: +82-2-598-1550 Fax: +82-2-598-1557 E-mail: composites@kscm.re.kr